Ethmozine: Package Insert / Prescribing Info
Package insert / product label
Generic name: moricizine hydrochloride
Dosage form: Tablets
Drug class: Group I antiarrhythmics
Medically reviewed by Drugs.com. Last updated on Mar 25, 2024.
On This Page
Ethmozine Description
Ethmozine® (moricizine hydrochloride) is an orally active antiarrhythmic drug available for administration in tablets containing 200 mg, 250 mg and 300 mg of moricizine hydrochloride. The chemical name of moricizine hydrochloride is 10-(3-morpholinopropionyl) phenothiazine-2-carbamic acid ethyl ester hydrochloride and the structural formula is represented as follows:
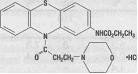
Moricizine hydrochloride is a white to tan crystalline powder, freely soluble in water and has a pKa of 6.4 (weak base). Ethmozine® tablets contain: lactose, magnesium stearate, microcrystalline cellulose, sodium starch glycolate and dyes (FD&C Blue 1, D&C Yellow 10 and FD&C Yellow 6 [200 mg tablet]; FD&C Yellow 6 and F&C Red 40 [250 mg tablet]; FD&C Blue 1 [300 mg tablet]).
Ethmozine - Clinical Pharmacology
Mechanism of Action
Ethmozine® is a Class I antiarrhythmic agent with potent local anesthetic activity and myocardial membrane stabilizing effects. Ethmozine® reduces the fast inward current carried by sodium ions.
In isolated dog Purkinje fibers, Ethmozine® shortens Phase II and III repolarization, resulting in a decreased action potential duration and effective refractory period. A dose-related decrease in the maximum rate of Phase 0 depolarization (Vmax) occurs without effect on maximum diastolic potential or action potential amplitude. The sinus node and atrial tissue of the dog are not affected.
Electrophysiology
Electrophysiology studies in patients with ventricular tachycardia have shown that Ethmozine®, at daily doses of 750 mg and 900 mg, prolongs atrioventricular conduction. Both AV nodal conduction time (AH interval) and His-Purkinje conduction time (HV interval) are prolonged by 10-13% and 21-26%, respectively. The PR interval is prolonged by 16-20% and the QRS by 7-18%. Prolongations of 2-5% in the corrected QT interval result from widening of the QRS interval, but there is shortening of the JT interval, indicating an absence of significant effect on ventricular repolarization. Intra-atrial conduction or atrial effective refractory periods are not consistently affected. In patients without sinus node dysfunction, Ethmozine® has minimal effects on sinus cycle length and sinus node recovery time. These effects may be significant in patients with sinus node dysfunction (see PRECAUTIONS: Electrocardiographic Changes/Conduction Abnormalities).
Hemodynamics
In patients with impaired left ventricular function, Ethmozine® has minimal effects on measurements of cardiac performance such as cardiac index, stroke volume index, pulmonary capillary wedge pressure, systemic or pulmonary vascular resistance or ejection fraction, either at rest or during exercise. Ethmozine® is associated with a small, but consistent increase in resting blood pressure and heart rate. Exercise tolerance in patients with ventricular arrhythmias is unaffected. In patients with a history of congestive heart failure or angina pectoris, exercise duration and rate-pressure product at maximal exercise are unchanged during Ethmozine® administration. Nonetheless, in some cases worsened heart failure in patients with severe underlying heart disease has been attributed to Ethmozine®.
Other Pharmacologic Effects
Although Ethmozine® is chemically related to the neuroleptic phenothiazines, it has no demonstrated central or peripheral dopaminergic activity in animals. Moreover, in patients on chronic Ethmozine®, serum prolactin levels did not increase.
Pharmacokinetics/Pharmacodynamics
The antiarrhythmic and electrophysiologic effects of Ethmozine® are not related in time course or intensity to plasma moricizine concentrations or to the concentrations of any identified metabolite, all of which have short (2-3 hours) half-lives. Following single doses of Ethmozine®, there is a prompt prolongation of the PR interval, which becomes normal within 2 hours, consistent with the rapid fall of plasma moricizine. JT interval shortening, however, peaks at about 6 hours and persists for at least 10 hours. Although an effect on VPD rates is seen within 2 hours after dosing, the full effect is seen after 10-14 hours and persists in full, when therapy is terminated, for more than 10 hours, after which the effect decays slowly, and is still substantial at 24 hours. This suggests either an unidentified, active, long half-life metabolite or a structural or functional "deep compartment" with slow entry from, and release to, the plasma. The following description of parent compound pharmacokinetics is therefore of uncertain relevance to clinical actions.
Following oral administration, Ethmozine® undergoes significant first-pass metabolism resulting in an absolute bioavailability of approximately 38%. Peak plasma concentrations of Ethmozine® are usually reached within 0.5-2 hours. Administration 30 minutes after a meal delays the rate of absorption, resulting in lower peak plasma concentrations, but the extent of absorption is not altered. Ethmozine® plasma levels are proportional to dose over the recommended therapeutic dose range.
The apparent volume of distribution after oral administration is very large (> 300 L) and is not significantly related to body weight. Ethmozine® is approximately 95% bound to human plasma proteins. This binding interaction is independent of Ethmozine® plasma concentration.
Ethmozine® undergoes extensive biotransformation. Less than 1% of orally administered Ethmozine® is excreted unchanged in the urine. There are at least 26 metabolites, but no single metabolite has been found to represent as much as 1 % of the administered dose, and as stated above, antiarrhythmic response has relatively slaw onset and offset. Two metabolites are pharmacologically active in at least one animal model: moricizine sulfoxide and phenothiazine-2-carbamic acid ethyl ester sulfoxide. Each of these metabolites represents a small percentage of the administered dose (<0.6%), is present in lower concentrations in the plasma than the parent drug, and has a plasma elimination half-life of approximately three hours.
Ethmozine® has been shown to induce its own metabolism. Average Ethmozine® plasma concentrations in patients decrease with multiple dosing. This decrease in plasma levels of parent drug does not appear to affect clinical outcome for patients receiving chronic Ethmozine® therapy. The plasma half-life of Ethmozine® is 1.5-3.5 hours (most values about 2 hours) following single or multiple oral doses in patients with ventricular ectopy. Approximately 56% of the administered dose is excreted in the feces and 39% is excreted in the urine. Some Ethmozine® is also recycled through enterohepatic circulation.
CLINICAL ACTIONS
Ethmozine® at daily doses of 600-900 mg produces a dose-related reduction in the occurrence of frequent ventricular premature depolarizations (VPDs) and reduces the incidence of nonsustained and sustained ventricular tachycardia (VT). In controlled clinical trials, Ethmozine® has been shown to have antiarrhythmic activity that is generally similar to that of disopyramide, propranolol and quinidine at the doses studied. In controlled and compassionate use programmed electrical stimulation studies (PES), Ethmozine® prevented the induction of sustained ventricular tachycardia in approximately 25% (19/75) of patients. In a post-marketing randomized comparative PES study, Ethmozine® had a response rate of approximately 12% (7/59). Activity of Ethmozine® is maintained during long-term use.
Ethmozine® is effective in treating ventricular arrhythmias in patients with and without organic heart disease. Ethmozine® may be effective in patients in whom other antiarrhythmic agents are ineffective, not tolerated and/or contraindicated.
Arrhythmia exacerbation or "rebound" is not noted following discontinuation of Ethmozine® therapy.
Indications and Usage for Ethmozine
Ethmozine® is indicated for the treatment of documented ventricular arrhythmias, such as sustained ventricular tachycardia, that, in the judgement of the physician are life-threatening. Because of the proarrhythmic effects of Ethmozine®, its use with lesser arrhythmias is generally not recommended. Treatment of patients with asymptomatic ventricular premature contractions should be avoided.
Initiation of Ethmozine® treatment, as with other antiarrhythmic agents used to treat life-threatening arrhythmias, should be carried out in the hospital. Antiarrhythmic drugs have not been shown to enhance survival in patients with ventricular arrhythmias.
Contraindications
Ethmozine® (moricizine hydrochloride) is contraindicated in patients with pre-existing second- or third-degree AV block and in patients with right bundle branch block when associated with left hemiblock (bifascicular block) unless a pacemaker is present. Ethmozine® is also contraindicated in the presence of cardiogenic shock or known hypersensitivity to the drug.
Warnings
Mortality
Ethmozine® was one of the three antiarrhythmic drugs included in the National Heart Lung and Blood Institute's (NHLBI) Cardiac Arrhythmia Suppression Trial (CAST I), a long-term, multi-center, randomized, double-blind study in patients with asymptomatic non-life-threatening ventricular arrhythmias who had had a myocardial infarction more than six days, but less than two years previously. An excessive mortality or non-fatal cardiac arrest rate was seen in patients treated with both of the Class IC agents included in the trial, which led to discontinuation of those two arms of the trial. The average duration of treatment with these agents was 10 months. The Ethmozine® and placebo arms of the trial were continued in the NHLBI-sponsored CAST II. In this randomized, double-blind trial, patients with asymptomatic, non-life-threatening ventricular arrhythmias who had had a myocardial infarction within 4 to 90 days and left ventricular ejection fraction ≤0.40 prior to enrollment were evaluated. The average duration of treatment with Ethmozine® in this study was 18 months. The study was discontinued because of the unlikely possibility of demonstrating a benefit toward improved survival with Ethmozine® and because of an evolving adverse trend after long-term treatment, although there was no statistical significance versus placebo.
The applicability of the CAST results to other populations (e.g., those without recent myocardial infarction) is uncertain. Considering the known proarrhythmic properties of Ethmozine® and the lack of evidence of improved survival for any antiarrhythmic drug in patients without life-threatening arrhythmias, the use of Ethmozine®, as well as other antiarrhythmic agents, should be reserved for patients with life-threatening ventricular arrhythmias.
Proarrhythmia
Like other antiarrhythmic drugs, Ethmozine® can provoke new rhythm disturbances or make existing arrhythmias worse. These proarrhythmic effects can range from an increase in the frequency of VPDs to the development of new or more severe ventricular tachycardia, e.g., tachycardia that is more sustained or more resistant to conversion to sinus rhythm, with potentially fatal consequences. It is often not possible to distinguish a proarrhythmic effect from the patient's underlying rhythm disorder, so that the occurrence rates given below must be considered approximations. Note also that drug-induced arrhythmias can generally be identified only when they occur early after starting the drug and when the rhythm can be identified, usually because the patient is being monitored. It is clear from the NIH sponsored CAST (Cardiac Arrhythmia Suppression Trial) that some antiarrhythmic drugs can cause increased sudden death mortality, presumably due to new arrhythmias or asystole that do not appear early after treatment but that represent a sustained increased risk.
Domestic pre-marketing trials included 1072 patients given Ethmozine®; 397 had baseline lethal arrhythmias (sustained VT or VF and non-sustained VT with hemodynamic symptoms) and 576 had potentially lethal arrhythmias (increased VPDs or NSVT in patients with known structural heart disease, ischemic heart disease, congestive heart failure or an LVEF <40% and/or Cl <2.01/min/m2). In this population there were 40 (3.7%) identified proarrhythmia events, 26 (2.5%) of which were serious, either fatal (6), new hemodynamically significant sustained VT or VF (4), new sustained VT that was not hemodynamically significant (11) or sustained VT that became syncopal/presyncopal when it had not been before (5). Proarrhythmic effects described as incessant ventricular tachycardia were observed in the post-marketing PES study and in post-marketing adverse event reports.
In general, serious proarrhythmic effects in the domestic pre-marketing trials were equally common in patients with more and less severe arrhythmias, 2.5% in the patients with baseline lethal arrhythmias vs. 2.8% in patients with potentially lethal arrhythmias, although the patients with serious effects were more likely to have a history of sustained VT (38% vs. 23%). In the post-marketing comparative PES study, patients treated with Ethmozine® (250-300 mg TID) had a proarrhythmia rate of 14% (8/59).
Five of the six fatal proarrhythmic events were in patients with baseline lethal arrhythmias; four had prior cardiac arrests. Rates and severity of proarrhythmic events were similar in patients given 600-900 mg of Ethmozine® per day and those given higher doses. Patients with proarrhythmic events were more likely than the overall population to have coronary artery disease (85% vs. 67%), history of acute myocardial infarction (75% vs. 53%), congestive heart failure (60% vs. 43%), and cardiomegaly (55% vs. 33%). All of the six proarrhythmic deaths were in patients with coronary artery disease; 5/6 each had documented acute myocardial infarction, congestive heart failure, and cardiomegaly.
Precautions
General:
Electrocardiographic Changes/Conduction Abnormalities
Ethmozine® slows AV nodal and intraventricular conduction, producing dose-related increases in the PR and QRS intervals. In clinical trials, the average increase in the PR interval was 12% and the QRS interval was 14%. Although the QTC interval was increased, this is wholly because of QRS prolongation; the JT interval is shortened, indicating the absence of significant slowing of ventricular repolarization. The degree of lengthening of PR and QRS intervals does not predict efficacy.
In controlled clinical trials and in open studies, the overall incidence of delayed ventricular conduction, including new bundle branch block pattern, was approximately 9.4%. In patients without baseline conduction abnormalities, the frequency of second-degree AV block was 0.2% and third-degree AV block did not occur. In patients with baseline conduction abnormalities, the frequencies of second-degree AV block and third-degree AV block were 0.9% and 1.4%, respectively.
Ethmozine® therapy was discontinued in 1.6% of patients due to electrocardiographic changes (0.6% due to sinus pause or asystole, 0.2% to AV block, 0.2% to junctional rhythm, 0.4% to intraventricular conduction delay, and 0.2% to wide QRS and/or PR interval).
In patients with pre-existing conduction abnormalities, Ethmozine® therapy should be initiated cautiously. If second- or third-degree AV block occurs, Ethmozine® therapy should be discontinued unless a ventricular pacemaker is in place. When changing the dose of Ethmozine® or adding concomitant medications which may also affect cardiac conduction, patients should be monitored electrocardiographically.
Hepatic Impairment
Patients with significant liver dysfunction have reduced plasma clearance and an increased half-life of Ethmozine®. Although the precise relationship of Ethmozine® levels to effect is not clear, patients with hepatic disease should be treated with lower doses and closely monitored for excessive pharmacological effects, including effects on ECG intervals, before dosage adjustment. Patients with severe liver disease should be administered Ethmozine® with particular care, if at all (see DOSAGE AND ADMINISTRATION).
Renal Impairment
Plasma levels of intact Ethmozine® are unchanged in hemodialysis patients, but a significant portion (39%) of Ethmozine® is metabolized and excreted in the urine. Although no identified active metabolite is known to increase in people with renal failure, metabolites of unrecognized importance could be affected. For this reason, Ethmozine® should be administered cautiously in patients with impaired renal function. Patients with significant renal dysfunction should be started on lower doses and monitored for excessive pharmacologic effects, including ECG intervals, before dosage adjustment (see DOSAGE AND ADMINISTRATION).
Congestive Heart Failure
Most patients with congestive heart failure have tolerated the recommended Ethmozine® daily doses without unusual toxicity or change in effect. Pharmacokinetic differences between Ethmozine® patients with and without congestive heart failure were not apparent (See Hepatic Impairment above). In some cases, worsened heart failure has been attributed to Ethmozine®. Patients with pre-existing heart failure should be monitored carefully when Ethmozine® is initiated.
Drug Interactions
No significant changes in serum digoxin levels or pharmacokinetics have been observed in patients or healthy subjects receiving concomitant Ethmozine® therapy. Concomitant use was associated with additive prolongation of the PR interval, but not with a significant increase in the rate of second- or third-degree AV block.
Concomitant administration of cimetidine resulted in a decrease in Ethmozine® clearance of 49% and a 1.4 fold increase in plasma levels in healthy subjects. During clinical trials, no significant changes in the efficacy or tolerance of Ethmozine® have been observed in patients receiving concomitant cimetidine therapy. Patients on cimetidine should have Ethmozine® therapy initiated at relatively low doses, not more than 600 mg/day. Patients should be monitored when concomitant cimetidine therapy is instituted or discontinued or when the Ethmozine® dose is changed.
Concomitant administration of beta blacker therapy did not reveal significant changes in overall electrocardiographic intervals in patients. In one controlled study, Ethmozine® (moricizine hydrochloride) and propranolol administered concomitantly produced a small additive increase in the PR interval.
Theophylline clearance and plasma half-life were significantly affected by multiple dose Ethmozine® administration when both conventional and sustained release theophylline were given to healthy subjects (clearance increased 44-66% and plasma half-life decreased 19-33%). Plasma theophylline levels should be monitored when concomitant Ethmozine® is initiated or discontinued.
Because of possible additive pharmacologic effects, caution is indicated when Ethmozine® is used with any drug that affects cardiac electrophysiology. Uncontrolled experience in patients indicates no serious adverse interaction during the concomitant use of Ethmozine® and diuretics, vasodilators, antihypertensive drugs, calcium channel blockers, beta blockers, angiotensin-converting enzyme inhibitors, or warfarin. Plasma warfarin levels, warfarin pharmacokinetics, and prothrombin times were unaffected during multiple dose Ethmozine® administration to young, healthy, male subjects in a controlled study. However, there are isolated reports of the need to either increase or decrease warfarin doses after initiation of Ethmozine®. Some patients who were taking warfarin with a stable prothrombin time experienced excessive prolongation of the prothrombin time following the initiation of Ethmozine®. In some cases, liver enzymes also were elevated. Bleeding or bruising may occur. When Ethmozine® is started or stopped in a patient stabilized on warfarin, more frequent prothrombin time monitoring is advisable.
Results from in vitro studies do not suggest alterations in Ethmozine® plasma protein binding in the presence of other highly plasma protein bound drugs.
CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY
In a 24-month mouse study in which Ethmozine® was administered in the feed at concentrations calculated to provide doses ranging up to 320 mg/kg/day, ovarian tubular adenomas and granulosa cell tumors were limited in occurrence to Ethmozine® treated animals. Although the findings were of borderline statistical significance, or not statistically significant, historical control data indicate that both of these tumors are uncommon in the strain of mouse studied.
In a 24-month mouse study in which Ethmozine® was administered by gavage to rats at doses of 25, 50 and 100 mg/kg/day, Zymbal's Gland Carcinoma was observed in one mid-dose and two high-dose males. This tumor appears to be uncommon in the strain of rat studied. Rats of both sexes showed a dose-related increase in hepatocellular cholangioma (also described as bile ductile cystadenoma or cystic hyperplasia) along with fatty metamorphosis, possibly due to disruption of hepatic choline utilization for phospholipid biosynthesis. The rat is known to be uniquely sensitive to alteration in choline metabolism.
Ethmozine® was not mutagenic when assayed for genotoxicity in in vitro bacterial (Ames test) and mammalian (Chinese hamster ovary/hypoxanthine-guanine phosphoribosyl transferase and sister chromatid exchange) cell systems or in in vivo mammalian systems (rate bone cytogenicity and mouse micronucleus).
A general reproduction and fertility study was conducted in rats at dose levels up to 6.7 times the maximum recommended human dose of 900 mg/day (based upon 50 kg human body weight) and revealed no evidence of impaired male or female fertility.
Pregnancy
Teratogenic Effects: Pregnancy Category B
Teratology studies have been performed with Ethmozine® in rats and in rabbits at doses up to 6.7 and 4.7 times the maximum recommended human daily dose, respectively, and have revealed no evidence of harm to the fetus. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, Ethmozine® should be used during pregnancy only if clearly needed.
Pregnancy-Nonteratogenic Effects:
In a study in which rats were dosed with Ethmozine® prior to mating, during mating and throughout gestation and lactation, dose levels 3.4 and 6.7 times the maximum recommended human daily dose produced a dose-related decrease in pup and maternal weight gain, possibly related to a larger litter size. In a study in which dosing was begun on Day 15 of gestation, Ethmozine®, at a level 6.7 times the maximum recommended human daily dose, produced a retardation in maternal weight gain but no effect on pup growth.
Nursing Mothers
Ethmozine® is secreted in the milk of laboratory animals and has been reported to be present in human milk. Because of the potential for serious adverse reactions in nursing infants from Ethmozine®, a decision should be made whether to discontinue the drug, taking into account the importance of the drug to the mother.
Adverse Reactions/Side Effects
The most serious adverse reaction reported for Ethmozine® is proarrhythmia (see WARNINGS). This occurred in 3.7% of 1072 patients with ventricular arrhythmias who received a wide range of doses under a variety of circumstances.
In addition to discontinuations because of proarrhythmias, in controlled clinical trials and in open studies, adverse reactions led to discontinuation of Ethmozine® in 7% of 1105 patients with ventricular and supraventricular arrhythmias, including 3.2% due to nausea, 1.6% due to ECG abnormalities (principally conduction defects, sinus pause, junctional rhythm, or AV block), 1% due to congestive heart failure and 0.3-0.4% due to dizziness, anxiety, drug fever, urinary retention, blurred vision, gastrointestinal upset, rash, and laboratory abnormalities.
The most frequently occurring adverse reactions in the 1072 patients (including all adverse experiences whether or not considered Ethmozine®-related by the investigator) were dizziness (15.1%), nausea (9.6%), headache (8.0%), fatigue (5.9%), palpitations (5.8%) and dyspnea (5.7%). Dizziness appears to be related to the size of each dose. In a comparison of 900 mg/day given at 450 mg b.i.d. or 300 mg t.i.d., more than 20% of patients experienced dizziness on the b.i.d. regimen vs. 12% on the t.i.d. regimen.
Adverse reactions reported by less than 5%, but in 2% or greater of the patients were: sustained ventricular tachycardia, hypesthesias, abdominal pain, dyspepsia, vomiting, sweating, cardiac chest pain, asthenia, nervousness, paresthesias, congestive heart failure, muscoloskeletal pain, diarrhea, dry mouth, cardiac death, sleep disorders and blurred vision.
Adverse reactions infrequently reported (in less than 2% of the patients) were:
Cardiovascular hypotension, hypertension, syncope, supraventricular arrhythmias (including atrial fibrillation/flutter), cardiac arrest, bradycardia, pulmonary embolism, myocardial infarction, vasodilation, cerebrovascular events, thrombophiebitis;
Nervous System tremor, anxiety, depression, euphoria, confusion, somnolence, agitation, seizure, coma, abnormal gait, hallucinations, nystagmus, diplopia, speech disorder, akathisia, loss of memory, ataxia, abnormal coordination, dyskinesia, vertigo, tinnitus;
Genitourinary urinary retention or frequency, dysuria, urinary incontinence, kidney pain, impotence, decreased libido;
Respiratory hyperventilation, apnea, asthma, pharyngitis, cough, sinusitis;
Gastrointestinal anorexia, bitter taste, dysphagia, flatulence, ileus;
Other drug fever, hypothermia, temperature intolerance, eye pain, rash, pruritus, dry skin, urticaria, swelling of the lips and tongue, perorbital edema.
During Ethmozine® therapy, two patients developed thrombocytopenia that may have been drug-related. Clinically significant elevations in liver function tests (bilirubin, serum transaminases) and jaundice consistent with hepatitis were rarely reported. Although a cause and effect relationship has not been established, caution is advised in patients who develop unexplained signs of hepatic dysfunction, and consideration should be given to discontinuing therapy.
Three patients developed rechallenge-confirmed drug fever, with one patient experiencing an elevation above 103°F (to 105°F, with rigors). Fevers occurred at about 2 weeks in 2 cases, and after 21 weeks in the third. Fevers resolved within 48 hours of discontinuation of moricizine.
Adverse reactions were generally similar in patients over 65 (n=375) and under 65 (n=697), although discontinuation of therapy for reasons other than proarrhythmia was more common in older patients (13.9% vs. 7.7%). Overall mortality was greater in older patients (9.3% vs. 3.9%), but those were not deaths attributed to treatment and the older patients had more serious underlying heart disease.
The following table compares the most common (occurrence in more than 2% of the patients) non-cardiac adverse reactions (i.e. drug-related or of unknown relationship) in controlled clinical trials during the first one to two weeks of therapy with Ethmozine®, quinidine, placebo, disopyramide or propranolol in patients with ventricular arrhythmias.
| Adverse Reactions | >2% | >2% | >2% | >5% | >5% | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Morcizine | Placebo | Quinidine | Disopyramide | Propranolol | ||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | |
| Total No. of Patients | 1072 | 618 | 110 | 31 | 24 | |||||
| Dizziness | 121 | 11.3 | 33 | 5.3 | 8 | 7.3 | - | 2 | 8.3 | |
| Nausea | 74 | 6.9 | 18 | 2.9 | 7 | 6.4 | 3 | 9.7 | - | |
| Headache | 62 | 5.8 | 27 | 4.4 | 4 | 16.7 | ||||
| Pain | 41 | 3.8 | 31 | 5.0 | 6 | 5.5 | 2 | 6.5 | ||
| Dyspnea | 41 | 3.8 | 22 | 3.6 | ||||||
| Hypesthesia | 40 | 3.7 | - | 3 | 2.7 | - | - | |||
| Fatigue | 33 | 3.1 | 16 | 2.6 | 6 | 5.5 | 2 | 6.5 | 3 | 12.5 |
| Vomiting | 22 | 2.1 | - | |||||||
| Dry Mouth | - | - | 11 | 35.5 | - | |||||
| Nervousness | - | - | - | 3 | 9.7 | - | ||||
| Blurred Vision | - | - | 3 | 2.7 | 2 | 6.5 | 3 | 12.5 | ||
| Diarrhea | - | - | 25 | 22.7 | - | - | ||||
| Constipation | - | - | - | 2 | 6.5 | - | ||||
| Somnolence | - | - | 2 | 8.3 | ||||||
| Urinary Retention | - | - | 4 | 12.9 | ||||||
Overdosage
Deaths have occurred after accidental or intentional overdosages of 2,250 and 10,000 mg of Ethmozine® (moricizine hydrochloride), respectively.
Signs, Symptoms and Laboratory Findings Associated with an Overdosage of Drug
Overdosage with Ethmozine® may produce emesis, lethargy, coma, syncope, hypotension, conduction disturbances, exacerbation of congestive heart failure, myocardial infarction, sinus arrest, arrhythmias (including junctional bradycardia, ventricular tachycardia, ventricular fibrillation and asystole), and respiratory failure.
Lethal Dose in Animals
Oral doses of Ethmozine® of about 200 mg/kg in dogs, 250 mg/kg in monkeys, 420 mg/kg in mice and 905 mg/kg in rats were lethal to about one-half of the animals exposed. Death was usually preceded by tremors, convulsions and respiratory depression.
Recommended General Treatment Procedures
A specific antidote for Ethmozine® has not been identified. In the event of overdosage, treatment should be supportive. Patients should be hospitalized and monitored for cardiac, respiratory and CNS changes. Advanced life support systems, including an intracardiac pacing catheter, should be provided where necessary. Acute overdosage should be treated with appropriate gastric evacuation, and with special care to avoid aspiration. Accidental introduction of Ethmozine® into the lungs of monkeys resulted in rapid arrhythmic death.
Ethmozine Dosage and Administration
The dosage of Ethmozine® must be individualized on the basis of antiarrhythmic response and tolerance. Clinical, cardiac rhythm monitoring, electrocardiogram intervals, exercise testing, and/or programmed electrical stimulation testing may be used to guide antiarrhythmic response and dosage adjustment. In general, the patients will be at high risk and should be hospitalized for the initiation of therapy (see INDICATIONS AND USAGE).
The usual adult dosage is between 600 and 900 mg per day, given every 8 hours in three equally divided doses. Within this range, the dosage can be adjusted as tolerated, in increments of 150 mg/day at 3-day intervals, until the desired effect is obtained. Patients with life-threatening arrhythmias who exhibit a beneficial response as judged by objective criteria (Holter monitoring, programmed electrical stimulation, exercise testing, etc.) can be maintained on chronic Ethmozine® therapy. As the antiarrhythmic effect of Ethmozine® persists for more than 12 hours, some patients whose arrhythmias are well-controlled on a Q8H regimen may be given the same total daily dose in a Q12H regimen to increase convenience and help assure compliance. When higher doses are used, patients may experience more dizziness and nausea on the Q12 hour regimen.
Patients with Hepatic Impairment
Patients with hepatic disease should be started at 600 mg/day or lower and monitored closely, including measurement of ECG intervals, before dosage adjustment.
Patients with Renal Impairment
Patients with significant renal dysfunction should be started at 600 mg/day or lower and monitored closely, including measurement of ECG intervals, before dosage adjustment.
Transfer to Ethmozine®
Recommendations for transferring patients from another antiarrhythmic to Ethmozine® can be given based on theoretical considerations. Previous antiarrhythmic therapy should be withdrawn for 1-2 plasma half-lives before starting Ethmozine® at the recommended dosages. In patients in whom withdrawal of a previous antiarrhythmic is likely to produce life-threatening arrhythmias, hospitalization is recommended.
| Transferred From | Start Ethmozine® |
|---|---|
| Quinidine, Disopyramide | 6-12 hours after last dose |
| Procainamide | 3-6 hours after last dose |
| Encainide, Propafenone, Tocainide, or Mexiletine | 8-12 hours after last dose |
| Flecainide | 12-24 hours after last dose |
How is Ethmozine supplied
Ethmozine® (moricizine hydrochloride) is available as oval, convex, film-coated tablets as follows:
| 200 mg (light green): | Bottles of 100 | (NDC 54092-046-01) |
| Hospital Unit Dose Carton of 100 | (NDC 54092-046-52) | |
| 250 mg (light orange): | Bottles of 100 | (NDC 54092-047-01) |
| Hospital Unit Dose Carton of 100 | (NDC 54092-047-52) | |
| 300 mg (light blue): | Bottles of 100 | (NDC 54092-048-01) |
| Hospital Unit Dose Carton of 100 | (NDC 54092-048-52) |
Store at 25°C (77°F) excursions permitted to 15-30°C (59-86°F) [See USP Controlled Room Temperature]
Shire
Manufactured for
Shire US Inc., 7900 Tanners Gate Drive, Suite 200, Florence, KY 41042 USA
1-800-828-2088, Made in USA
©2001 Shire US Inc.
U.S. Patent 3,864,487
046 0117 006 / Rev. 2/02
6322-04 / February 2002
| ETHMOZINE
moricizine hydrochloride tablet |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| ETHMOZINE
moricizine hydrochloride tablet |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| ETHMOZINE
moricizine hydrochloride tablet |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - Shire US Inc. |
More about Ethmozine (moricizine)
- Check interactions
- Compare alternatives
- Reviews (1)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: group I antiarrhythmics
